The Menu
As surface science has increasingly moved to the nanoscale over the past decade, it has entered a realm that is also accessible to highly accurate computational approaches. Simulations have become a key partner in many studies, with the interplay between theory and experiment being the critical element in real understanding. In this overview presentation, I will try to introduce the most popular tools used in computational surface science, along with examples highlighting their interaction with experiment. I will also show examples where common methods fail, and point out the consequences of some "short-cuts" often hidden in the technical details.
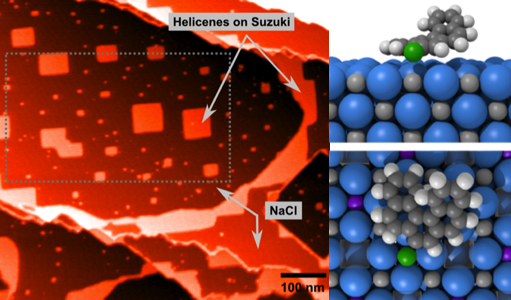
The family of scanning probe microscopy (SPM) methods employing forces for studying surface properties has developed over the last 25 years into many versatile tools in surface physics and chemistry, as well as in nanoscience and nanotechnology. In particular, two of the most commonly used techniques, non- contact atomic force microscopy (nc-AFM) and Kelvin probe force microscopy (KPFM), have been applied to study the surfaces of insulators and thin films with atomic or nanoscale resolution. A major strength of these techniques is their lack of material restrictions, and many of the breakthrough results in high-resolution imaging have been achieved on surfaces inaccessible to other imaging techniques [1]. In this work we initially use the NaCl (001) as a benchmark insulating surface linking comprehensive SPM studies using various modes. In general we link first principles and atomistic simulations directly into realistic simulators, taking account of the nature of the tip and surface in experiments and including the fundamental components of the experimental setup.
NaCl surfaces are prepared by cleavage, resulting in a rich selection of defects on the surface. By combining experimental and simulated images using conventional nc- AFM mode with KPFM [2], and nanoparticle manipulation studies, we characterize the key defects on the surface. We further explore doping of NaCl with divalent ions to precipitate nano-islands of impurities and vacancies on the surface, offering immediate characterization of nc-AFM atomically resolved images [3], as well as nanocluster and molecular templating. We finally compare the results with more complex defect structures and chemistry we have observed in atomic scale studies of oxide systems [4].
[1] C. Barth et al., Advanced Materials 23 477 (2011).
[2] Laurent Nony et al., Physical Review Letters 103 036802 (2009).
[3] Adam S. Foster et al., Physical Review Letters 102 256103 (2009).
[4] M. Rasmussen et al., Physical Review Letters 107 036102 (2011).
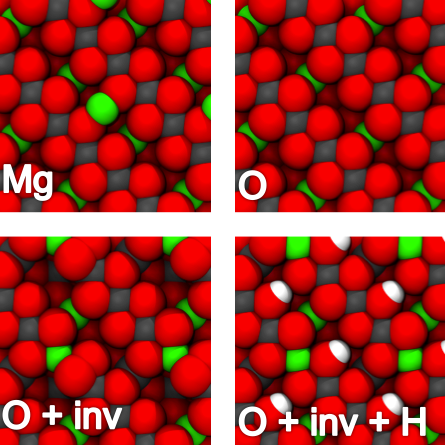
Atomic defects have an overriding effect on physical and chemical properties of oxide materials, and many technologies rely on detailed control of atomic impurities, promoters, or dopants, such as in gate oxides, optics, or (photo)catalysts of electronic states pinned at defects. Scanning probe microscopy (SPM) techniques allow characterization of these defects at the atomic scale, but interpretation remains challenging, particularly when only one signal is measured. In this work, we show how a combination of SPM and other experimental techniques with detailed Density Functional Theory (DFT) simulations can provide atomic scale characterization of defects and defect processes in two very different oxides.
As a demonstration of the approach, we use an interplay between simultaneously recorded non-contact atomic force microscopy (AFM) and scanning tunneling microscopy (STM) images [1] and DFT simulations, to reveal the location of single hydrogen species in the surface and subsurface layers of rutile TiO2. Subsurface hydrogen atoms are found to reside in a stable interstitial site as subsurface OH groups detectable in scanning tunneling microscopy as a characteristic electronic state, but imperceptible to atomic force microscopy [2].
For the insulating ternary metal oxide MgAl2O4 (spinel), we combine NC-AFM, Surface X- ray Diffraction (SXRD) experiments and DFT calculations to reveal the detailed atomic-scale structure of the polar (100) surface [3]. Contrary to earlier predictions, the surface is terminated by an Al and O-rich structure with a thermodynamically favored amount of Mg-Al antisite defects. These are low-density defects in the bulk of MgAl2O4, but become a thermodynamically stable and integral part of the surface.
[1] Georg H. Enevoldsen et al., Phys. Rev. B 78 045416 (2008).
[2] Georg H. Enevoldsen et al., Phys. Rev. Lett. 102 136103 (2009); Phys. Rev. Lett. 104 119604 (2010).
[3] M. Rasmussen et al., Phys. Rev. Lett. 107 036102 (2011).
It is possible to combine non-contact Atomic Force Microscopy (nc-AFM) and Kelvin Probe Force Microscopy (KPFM) techniques in a single measurement to obtain a detailed analysis of the topography and local work function of a surface. However, as the imaging AFM tip has a fundamental influence on the measured signals, the methods also provide information on the properties of the tip itself. In this study, we first demonstrate the basic contrast formation in nc-AFM and KPFM on MgO (001) thin films supported on Ag (001) [1]. It is shown that the topography contrast depends strongly on the charge state or polarity of the AFM tip due to coupling of the tip charge to the surface dipole present on MgO islands embedded in or supported by the Ag substrate. Furthermore, we show that the average Kelvin voltage in KPFM also allows one to observe the charge state of the tip. Both positively and negatively charged or polarized tips can be distinguished from neutral tips and from each other, as the mean Kelvin voltage can shift by tens of millielectronvolts when a tip change affecting the tip charge distribution occurs. The experimental topography measurements are compared to first principles calculations in order to demonstrate how the charge distribution in the tip affects the contrast formation. For the Kelvin signal, a simple analytic result is presented and compared to both experimental results and atomistic simulations, with excellent agreement between the different descriptions. [2, 3]
[1] M. Bieletzki et al., Phys. Chem. Chem. Phys. 12 3203 (2010).
[2] C. Barth et al., New J. Phys. 12 093024 (2010).
[3] T. Hynninen et al., E-J. Surf. Sci. Nanotech. 9 6 (2011).
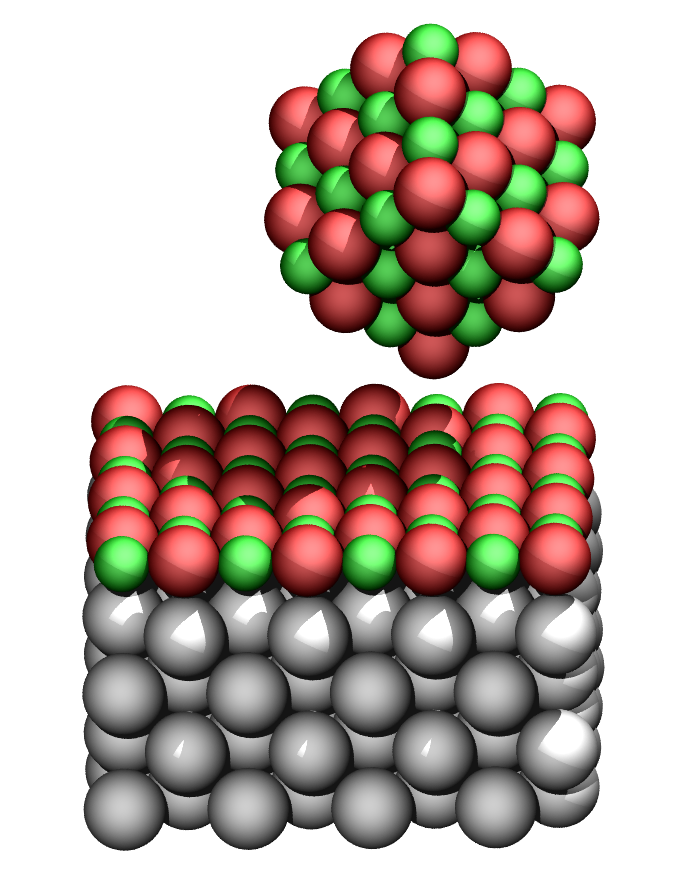
Controlling structures at the atomic level is the ultimate challenge in nanofabrication. Although arranging structures atom-by-atom is interest in fundamental surface physics and chemistry, for many applications manipulation of larger building blocks is more relevant. With respect to metallic clusters, interest stems from catalysis where clusters promote many catalytic reactions, but also from nanotechnology where clusters can be used as building blocks for nano-electronic devices. In tribology, clusters can even serve as units for probing the friction at the nanoscale.
We study Au cluster manipulation on NaCl (001), a prototype system, by both experiments and simulations. On the NaCl surface, gold atoms tend to aggregate at steps forming clusters which can then be manipulated with a non-contact atomic force microscope. Interestingly, the movement of clusters is seen to be strongly anisotropic, preferring [110] surface directions. By calculating the energy landscape of Au clusters on the surface using density-functional theory it is found that the anisotropy in minimum energy paths depends on the type of vacancy the clusters attach to, demonstrating how the manipulation behavior of metallic clusters on insulators can be dominated by the interaction between the clusters and the defects found on the surface.

When solid ice melts due to high pressure and then resolidifies once the pressure is lifted, the phenomenon is called regelation. A well-known example is the experiment where a wire under load passes through a solid block of ice without cutting the solid [1]. In nature, regelation plays a part, e.g., in glacial flows, as the melting point of water at the bottom of a glacier may be substantially lowered due to the elevated pressure, allowing the ice to deform around obstacles on its path [2].
We examine a nanowire regelation process, allowing us to study ice regelation and bulk friction on the molecular level. This is done by simulating a nanowire, encased in ice, being pushed through the solid by an external force. We use a molecular dynamics scheme where water is described using a geometric coarse-grained approach: the modified 3D Mercedes-Benz model [3]. This model is known to correctly yield a negative slope for the solid-liquid coexistence curve, i.e., it results in ice melting under increased pressure. We show that the method also captures the regelation of water back to ice. For the motion of the wire through the solid, we find a transition from a pinned state to stick-slip motion and steady flow as the driving force increases. The onset of movement is found to be hysteretic only if the surface of the wire is hydrophobic. [4]
[1] J. Thomson Bottomley, Nature 5 185 (1872).
[2] Dash et al., Rev. Mod. Phys. 78 695 (2006).
[3] C. L. Dias et al., J. Chem. Phys. 131 054505 (2009).
[4] T. Hynninen et al. Phys. Rev. Lett. 105 086102 (2010).

The atomic force microscope (AFM) can be operated in liquid environments, where it can be used to image flat surfaces with high resolution, study the structure of hydration layers, or image and manipulate biological systems on the nanoscale [1]. However, in all these applications, the tip-surface interaction mechanism is more complicated than in vacuum or air, and therefore generally not well understood.
In order to better understand AFM imaging in water, we chose calcite as a model system. The calcite (10-14) surface has been studied extensively, because of its important role in bio-mineralization, industrial applications, and as substrate for biomolecule growth and manipulation. Although recent atomically resolved non-contact AFM images of calcite in water [2] resemble those obtained in UHV [3], the imaging mechanism in liquid is more complicated [4], due to the presence of hydration layers around the tip and above the surface. Displacing these confined water molecules leads to significant entropic contributions to the force on the AFM tip.
In this work, we study the AFM imaging mechanism of calcite in water using molecular dynamics simulations with empirical atomistic interaction potentials. The system, consisting of several layers of surface material, water, and the tip apex represented by a nanocluster, is simulated at constant volume and room temperature. We carry out many individual simulations, in each of which the position of the tip above the surface is constrained by a harmonic potential. We then use the 'umbrella sampling' scheme to compute a continuous free energy profile of the system, as a function of the tip-surface separation. The derivative of this free energy with respect to the tip-surface separation is the best estimate for the force acting on the AFM tip, as the important entropic contributions from interactions with hydration layer water molecules are correctly taken into account in the free energy calculation.
The force-distance curves obtained with this procedure can be directly compared to force spectroscopy data from experiment. We also use a 'virtual AFM' tool, recently developed in our group, to simulate AFM images based on these force-distance curves. This enables us to fully understand the imaging mechanism, and to interpret the contrasts observed in high resolution images.
[1] D. J. Müller and Y. F. Dufrêne, Nature Nanotechnology 3 261 (2008).
[2] S. Rode et al., Langmuir 25 2850 (2009).
[3] J. Schütte et al., Langmuir 26 8295 (2010).
[4] M. Watkins and A. L. Shluger, Phys. Rev. Lett. 105 196101 (2010).
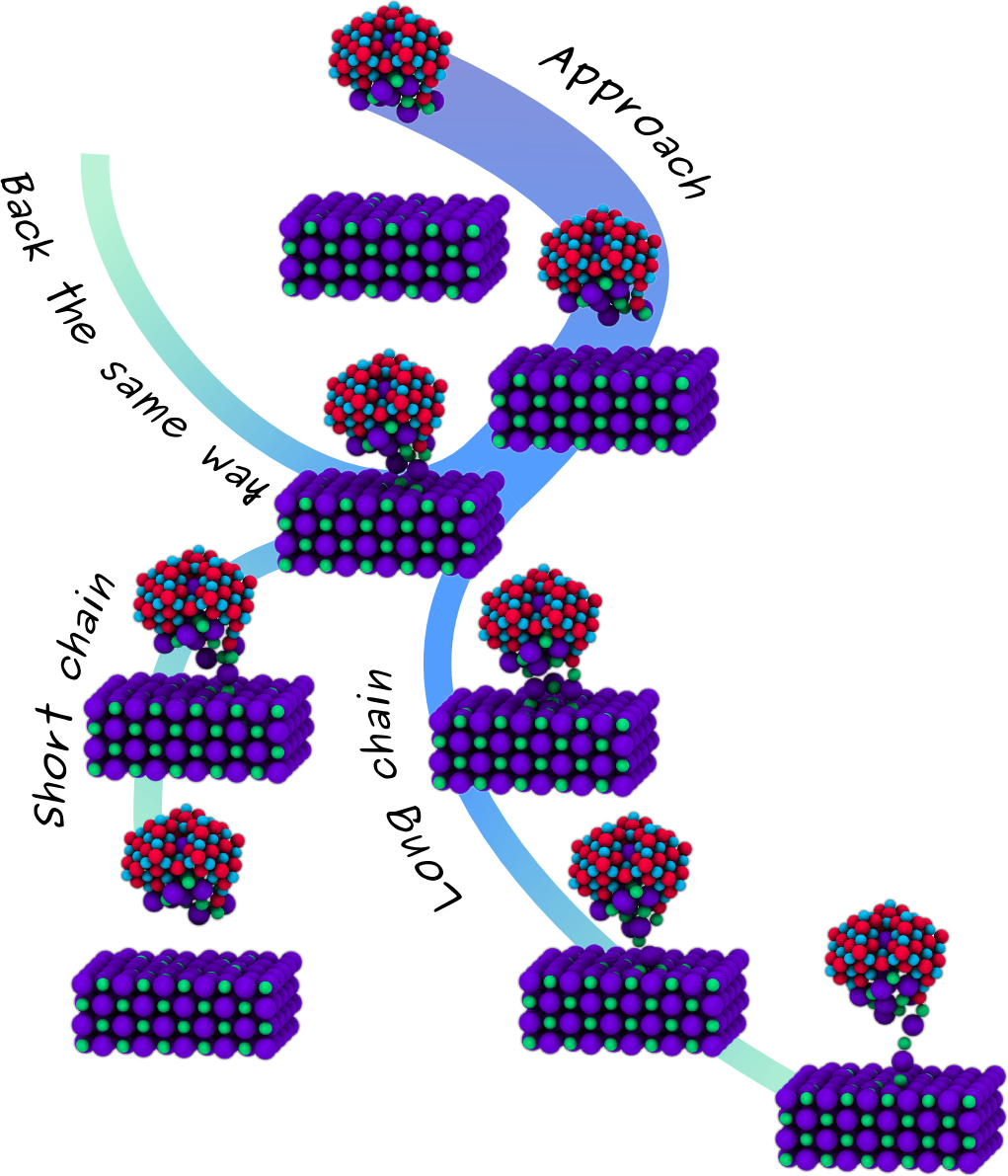
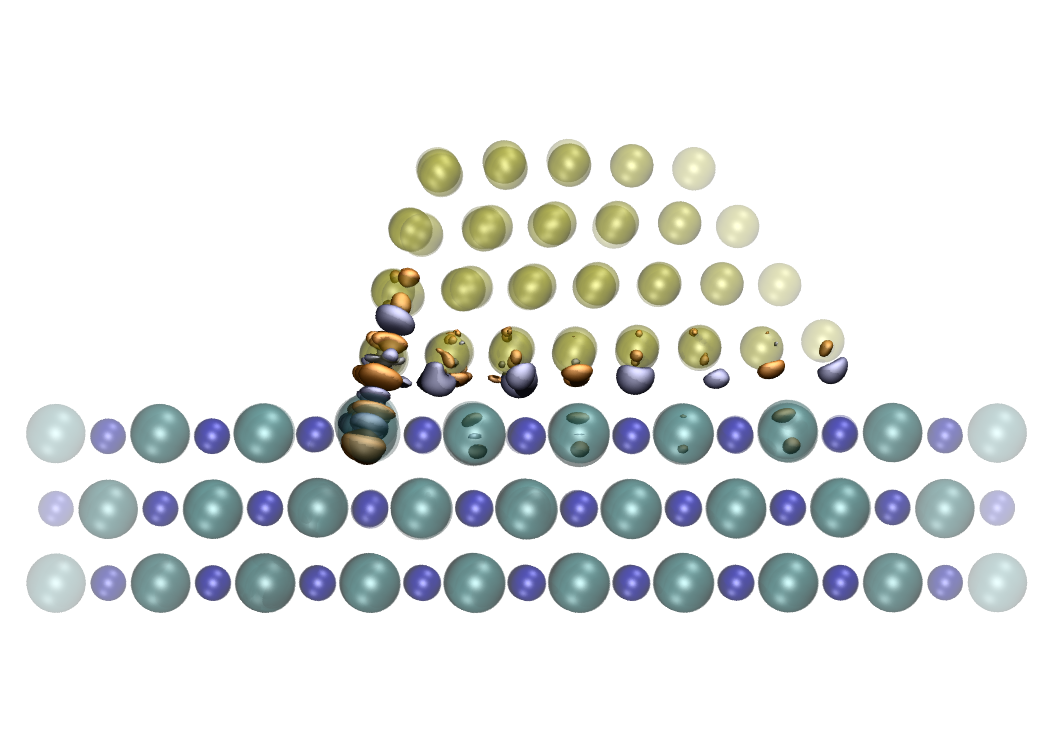
NC-AFM is an invaluable tool when probing surfaces, providing atomic resolution topography and detailed 3D force maps. As the tip scans the surface, the feedback gain signal associated to the energy dissipated in the oscillation cycles is also recorded, and this often differs from conventional topography images, showing different contrast patterns, periodicity and tip-surface distance dependence. Although many different surfaces have been studied with atomic resolution, a routine interpretation of these measurement has yet to be found, since the atomic processes at the surface responsible for energy dissipation are not well understood and the influence of the tip is not clear. In order to understand better the role of the tip in measured dissipation, we carried out extensive atomistic calculations using a wide variety of different tip materials and structures, and a simple NaCl (100) surface. After studying simple processes with quasi-static calculations [1], testing several tip geometries and materials [2], we are now investigating the atomic-scale dissipative processes using GPU accelerated finite temperature molecular dynamics (MD) simulations, and comparing directly to static force spectroscopy (SFS) and dynamic force spectroscopy (DFS) experiments. Ideal NaCl tips are too stable and do not give dissipation although in presence of defects at the apex, the system shows reversible reconstructions involving the tip and the surface, giving an average dissipation up to 0.02 eV/cycle, smaller than experimentally seen. MgO tips are even more stable but the oxygen's charge at the apex makes the surface unstable leading to formation of atomic chain and, consequently, irreversible surface alteration, which is again in contrast with the experiment, showing no decoration and stable operation. Finally we focused our efforts on simulating the experiment from the very beginning, where an oxidized tip indents the surface adsorbing a NaCl nanocluster at the apex. The average dissipation we calculate with this tip is in a quantitative agreement with DFS data and the distance dependence is also reproduced. We found that dissipation comes from stochastic formation of atomic chains of different lengths.
[1] L. N. Kantorovich and T. Trevethan, Phys. Rev. Lett. 93 236102 (2004).
[2] F. Federici Canova and A. S. Foster, Nanotechnology 22 045702 (2011).
